PP. 1-18
1Hariom Nagar* 1Renu Vyas, 2Shiv Alvera,
1School of Applied Sciences, Suresh Gyan Vihar University, Jaipur-302017
2Department of Chemical Science, Indian Institute of Science Education and Research Mohali, Punjab-140306
e-mail: hariomnagariitr@gmail.com; renuvyas6@gmail.com; Kumalvera@gmail.com
ABSTRACT
Orciprenaline is a moderately selective β-adrenergic receptor agonist and used in the treatment of asthma. Generally, the (S)-( ̶ )-enantiomer of β-blockers is pharmacologically effective, show about 50–500-fold higher activities than its (R)-( ̶ )-enantiomer. Diastereomers of orciprenaline were synthesized using a chiral derivatizing reagents that was prepared by substituting one fluorine atom in 1,5-difluoro-2,4-dinitrobenzene with chirally pure L-amino acid; L-Phenylalanine. Characterization of the chiral derivatizing reagent was made by using UV, IR, CHN, and 1H NMR. Diastereomers were synthesized under microwave irradiation 45 s at 80% (of 800W) and also by stirring for 50 min at 45 °C. Furthermore, the diastereomers were separated by high-performance liquid chromatography in reversed phase condition on a C18 column with detection at 340 nm. Acetonitrile and aqueous trifluoroacetic acid (TFA) were used as the mobile phase components. The conditions of derivatization and chromatographic separation were optimized to achieve good results. The method validation was made for accuracy, precision, limit of detection and limit of quantification.
KEYWORDS Orciprenaline, 1,5-Difluoro-2,4-dinitrobenzene, Enantioseparation, Liquid Chromatography,
INTRODUCTION
Orciprenaline (Orc) is a moderately selective β2-adrenergic receptor agonist and a bronchodilator used in the treatment of asthma. Generally, the (S)-(−)-enantiomer of β-blockers is pharmacologically effective, than its (R)- form and shows about 50–500 fold higher activities (Lee and William 1990). But in most cases β-blockers are administered as racemic mixtures.
Many clinical pharmacologists and biologists tend to deal the drug with mixtures of isomers as if one compound were involved and also a physician, presented with such a drug under a brand name is unaware of isomers and generally he then makes mistakes (Ariëns 1984). It is mostly because of non availability of single enantiomer as in enantiomerically purified form. In most of cases the undesired enantiomer can have side effects or even toxic effects. So it is prime importance and challenging task to determine optical purity of the drugs and if it is not enantiomerically pure then to resolve racemic mixtures in to its different enantiomers.
In literature, for separation of such drugs (β-blockers) there have been developed and used two approaches one is direct (no any type of chemical derivatization of enantiomers) and other is indirect approach (derivatization of enantiomers with chiral derivatizing reagent (CDR) to form their diastereomers) to achieve enantiomeric separation of β-blockers using the high performance liquid chromatographic (HPLC) techniques. HPLC techniques have been also utilised previously via indirect method for enantiomeric separation of various active pharmaceuticals; amino acids (Bhushan and Nagar 2013) and selenomethionine (Bhushan and Nagar 2014a) using CDRs based on (S)-naproxen moiety, isoxsuprine (Bhushan and Nagar 2015) and selenomethionine (Nagar and Bhushan 2014) using CDRs based on difluorodinitrobenzene moiety.
In literature, certain chiral stationary phases (CSPs) have been used for direct separation of enantiomers of Orc are; sulfobutyl ether β-cyclodextrin (SBECD) (Ngim et al. 2012), methyl-β-cyclodextrin (met-β-CD) (Amini et al. 2000) and sulfated-β-cyclodextrin (S-β-CD) (Yang et al. 2005) as chiral selector on C18 column, by direct approach in HPLC. L-Glutamic acid was used as chiral inducing reagent (CIR) for resolution of enantiomers of Orc (Bhushan and Nagar 2015) by direct approach in thin layer chromatography (TLC).
Besides above literature, Bhushan and Nagar (2014b) have been carried out enantioseparation of Orc by using 1,5-difluoro-2,4-dinitrobenzene (DFDNB) based chiral derivatizing reagents (CDRs) having amino acid and amines as chiral moiety.
DFDNB namely as Sanger’s reagent a bifunctional variant was first time used for cross linking of proteins such as wool, silk and insulin (Zahn and Meienhofer 1958). Marfey (1984) carried out the reaction of DFDNB with L-alanine amide and formed its chiral variant namely 1-fluoro-2,4-dinitrophenyl-L-alaninamide (FDNP-L-Ala-NH2, Marfey’s reagent), in this reaction one fluorine atom in DFDNB was substituted with L-alanine amide. This chiral derivatizing reagent (CDR) was used for HPLC enantioseparation of five DL-amino acids. DFDNB based CDRs have also been used in Bhushan laboratory for the separation of β-blockers (Bhushan and Tanwar 2008; 2009; Bhushan and Dixit 2012) other than the Orc, α-amino acids (Bhushan and Kumar 2008) and α-amino alcohols (Bhushan and Kumar 2009a). There have appeared certain review articles on use of Marfey’s reagent (B’Hymer et al. 2003; Bhushan and Brückner 2004; 2011) as CDR.
In view of the characteristics of DFDNB, the literature cited above and the references cited therein on the synthesis of DFDNB based CDRs, L-Phe was chosen as chiral amino acid for synthesis of DFDNB based CDR because the non bonding electron pair of amino group of L-Phe would show resonance by direct conjugation, with the para positioned –NO2 group and would be expected to further increase molar absorptivity of dinitrobenzene moiety. The chiral auxiliary in this CDR contain –CH2C6H5 as the bulky hydrophobic group which enhance the hydrophobicity of the CDR and are thus expected to influence the separation.
The CDR was used for liquid chromatographic enantioseparation of Orc compound since there are no reports on its enantioseparation using this DFDNB based CDR. The method was validated for linearity, accuracy, limit of detection (LOD) and limit of quantification (LOQ). To the best of the authors’ knowledge, this is the first report on use of this CDR for enantioseparation of Orc compound (Fig. 1).
EXPERIMENTAL
Chemicals and Reagents
1,5-Difluoro-2,4-dinitrobenzene, L-Phenylalanine (L-Phe), Orciprenaline sulphate as Alupent tablets (Zydus Healthcare, East Sikkim, India) was obtained from the local market. Trifluoroacetic acid (TFA), phosphoric acid (H3PO4), triethylamine (TEA), sodium hydrogen carbonate (NaHCO3) and hydrochloric acid (HCl) of analytical reagent grade, acetonitrile (MeCN) and methanol (MeOH) of HPLC grade were obtained from E. Merck (Mumbai, India). Aqueous–TFA (0.1%) and TEAP (50 mM) buffers were prepared in Millipore water.
Apparatus
The C18 column (Promosil, 250 × 4.6 mm I.D., 5 μm) was from younglin. HPLC consisting of a 10 mL pump head, vaccum degasser, Acme UV/Vis detector, younglin manual injection valve and Autochro-3000 operating software was from younglin (Gyeonggi-do, korea). UV-1601 spectrophotometer (Shimadzu), FT-IR spectrometer 1600 (Perkin-Elmer, USA), NMR spectrometer 500 MHz (Bruker, Germany), pH meter Cyberscan 510 (Singapore) and Microwave-Multiwave 3000 (800W, Perkin-Elmer, USA) equipments were also used for experimental studies.
Extraction of Orc from Commercial Tablets
Orc was extracted from commercially available “Alupent tablets” in local market. The extraction process was carried out as per the method described in literature (Bhushan and Nagar 2014b). The purity of the product was confirmed by determination of its melting point and UV absorption (λmax) spectra which were matching with literature reports. The recovery of compound was found in order of 93–95% of the quantities reported on the commercial labels. The purified compound was used as racemic standard.
Preparation of Stock Solutions
Solutions of NaHCO3 (1 M) and HCl (2 M) were prepared in purified water. Stock solution of racemic Orc was prepared by dissolving appropriate amount in 1 M NaHCO3 and used for
derivatization reactions. Solutions and samples were filtered by using 0.45 μm filters. Acetone was used as solvent to prepare solutions of CDR (25 mM) and then CDR stored in refrigerator at 0–4 °C for further uses.
Synthesis of CDR
Due to sensitivity of the reagent synthetic procedures were protected from light. The CDR was synthesized by introducing optically pure L-Phe via substitution of one fluorine atom in DFDNB as per method described in literature (Bhushan and Kumar 2009b). These were characterized and stored at 4 °C. A representative procedure for synthesis of CDR is given below. Following solutions were prepared.
- 1 M NaHCO3 in distilled water
- 1 M solution of L-Phe in (i), and
- 2 M solution of DFDNB in acetone-water (6:4, v/v); it was maintained at 0–4 oC
Synthesis of CDR (1-Fluoro-2,4-dinitrophenyl-L-phenylalanine; FDNP-L-Phe): 20 mL of (ii) was added to 20 mL of (iii)) with constant stirring, in a round bottom flask. The reaction mixture was constantly stirred at 40 °C; after 1 h, the resulting solution was kept at room temperature for 10 min and then 40 mL water was added to it. Further addition of 20 mL HCl (1 M) yielded yellow crystals; these were filtered and washed with cold water. The crystals were dehydrated in desiccators over P2O5.
The structure of the synthesized CDR (FDNP-L-Phe) is shown in Fig. 2 and its characterization data are in agreement with earlier reports (Bhushan and Kumar 2009b).
Synthesis of Diastereomers
The diastereomers of Orc with FDNP-L-Phe were synthesized according to the literature report (Bhushan and Nagar 2014b) using MW irradiation as well as conventional heating.
Following solutions were prepared for the purpose of synthesis of diastereomers.
- 1 M NaHCO3 in distilled water
- 50 mM solution of Orc in (iv), by dissolving 10.56 mg of Orc in 1 mL of (iv).
- 25 mM solution of FDNP-L-Phe in acetone by dissolving 8.73 mg of FDNP-L-Phe in 1 mL acetone; it was maintained at 0–4 o
Solution of Orc (as at (v) above) was added to the solution of FDNP-L-Phe (as at (vi) above) in a teflon tube. Following reaction conditions were tried to obtain an optimum reaction yield. Orc and FDNP-L-Phe were allowed to react in different ratios such as, 1:1, 1:1.5, 1:1.7 and 1:2. The reaction mixtures were microwave-irradiated in oven for 30, 40, 45, 50, 55, 60 and 70 s at each of the three different power settings, viz, 70, 80 and 90%. Separate sets of reaction mixture were incubated at 40, 45 and 50 °C with constant stirring for 40, 50, 60, 70 and 80 min (at each temperature). The scheme for synthesis of daistereomers by the reaction of CDR with (R/S)-Orc is given in Fig. 2. The formed daistereomers were processed through the HPLC experiment, the peak areas of each pair of the daistereomers obtained, were taken as a diagnosis for the completion of derivatization reactions of formation of the diastereomers and samples were taken as standards for separation.
A 10 μL of resulting solution of daistereomers was diluted to 100 μL with MeCN, degassed and filtered. Now 20 μL of it was injected onto the column through injector.
HPLC
Following mobile phases were tried;
Mobile phase 1, MeCN with TEAP buffer (10 mM) in linear gradient (30 to 80, 30 to 65, 35 to 60, 25 to 60, 25 to 70, 20 to 60, 20 to 70 and 10 to 90%) in 45 min run.
Mobile phase 2, MeCN with TFA buffer (0.1%) in linear gradient (30 to 80, 30 to 65, 35 to 60, 25 to 60, 25 to 70, 20 to 60, 20 to 70 and 10 to 90%) in 45 min run.
Mobile phase 3, MeOH with TFA buffer (0.1%) in linear gradient (30 to 80, 30 to 65, 35 to 60, 25 to 60, 25 to 70, 20 to 60, 20 to 70 and 10 to 90%) in 45 min run.
Chromatographic runs were carried out and optimized the conditions by varying concentration of buffer (TEAP, 5 to 20 mM and TFA 0.05% to 0.15%), pH (in the range 2.0 to 5.5), and flow rate (0.5 to 1.5 mL/min) of mobile phase. Before using, the mobile phases were filtered through a 0.45µm filter and degassed by sonication and by passing nitrogen.
On increasing the concentration of TFA from 0.05 to 0.10%, an increase in separation factor was observed. There was no significant increase in separation factor when its concentration was increased from 0.10 to 0.15%. High concentration of TFA, due to its strong acidic nature could be harmful to the column; therefore, 0.10% TFA was taken as the optimized concentration. A decrease in flow rate from 1 to 0.5 mL/min resulted in an increase in retention time with slight broadening of peaks while on the other hand, an increase in flow rate from 1 to 1.5 mL/min resulted in decrease of retention time and Δt.
Method Validation
Method was validated using diastereomers of Orc prepared with CDR (FDNP-L-Phe) according to ICH guidelines (ICH 1996). The peak areas were plotted against concentrations over the range 20 to 100 ng mL-1 to obtain the calibration curve and, slope and correlation coefficients were determined by using regression equation.
RESULTS AND DISCUSSION
Diastereomers
DFDNB is very good moiety in terms of detection and reactivity to form its derivatives. This is due to chromophoric effect of nitro group in DFDNB which enhances molar absorptivity while its strong electron withdrawing nature (-M/-R and –I effect) facilitates nucleophilic substitution of one of the fluorine atoms with L-Phe to yield the CDR (FDNP-L-Phe). Furthermore, a pair of diastereomers of good molar absorptivity was produced by substitution of remaining fluorine in CDR (FDNP-L-Phe) with chiral analyte. Better yield was obtained in the following optimised conditions: CDR (FDNP-L-Phe) and Orc are taken in the mole ratio of 1.7:1; heating the reaction mixture at 45 °C in an incubator for 50 min with constant stirring for conventional heating while irradiation of mixture for 45 s (at 80% of 800 W) under microwave condition in a separate set. The reaction was terminated by addition of HCl (2 M, 20 μL) after cooling to room temperature. Synthesis of diastereomers of Orc with FDNP-L-Phe was performed under the said optimized conditions. The 1.7 fold molar excess of CDR was successful to prevent kinetic resolution. The two diastereomers formed are of the type [(R)-L]-, and [(S)-L]- where the first letter refers to the configuration of the analyte (Orc ) while the second letter to that of the L-Phe of the CDR (FDNP-L-Phe).
HPLC
The data for resolution (RS) and separation factor (α) for separation of diastereomers are; 11.76 and 1.12, respectively. Sharp peaks were observed with the mobile phase under gradient conditions with MeCN used as organic modifier in mobile phase 1 and 2 while with MeOH in mobile phase 3 broader peaks were obtained with larger retention time. This is due to lower dielectric constant (33 D) and higher viscosity (0.59 cP at 25 °C) of MeOH in comparison to acetonitrile having dielectric constant (37.5 D) and viscosity (0.343 cP at 25 °C). With TFA buffer (mobile phase 2) sharper peaks were observed in comparison to those obtained using TEAP buffer (mobile phase 1). Thus, the mobile phase 2, in which MeCN used as organic modifier and 0.10% TFA as buffer, with gradient elution (30 to 65% of MeCN) in 45 min at a flow rate of 1.0 mL/min and detection at 340 nm was found successful for separation of daistereomers of Orc. The chromatogram showing separation of diastereomers of the Orc prepared with CDR (FDNP-L-Phe) is given in Fig. 3.
In HPLC run the peak areas obtained for corresponding daistereomers in each change of the above mentioned derivatization conditions, were calculated by system software; these values taken as a measure of completion of reaction and yield of derivatization. The characterization and chromatographic data of diastereomers synthesized in the two conditions (using conventional heating and microwave irradiation (MWI)) were found to be identical.
Comparison with literature reports
In the present study, derivatization of Orc using DFDNB based CDR (FDNP-L-Phe) in which L-Phe was introduced as chiral auxiliary under MWI required less time (45 s at 80% power of 800 W). Diastereomers of Orc have been separated with resolution (Rs) 11.76 which is better as compared to the resolution reported in literature (Table 1) where certain CSPs, and CDRs used in HPLC and CIR used in TLC.
Separation mechanism
Certain literature reports (Marfey 1984; Bhushan et al. 2009; Fujii et al. 1997) have been explained the mechanism for HPLC separation of diastereomers of certain amino acids, prepared with DFDNB based CDRs, along with explanation for difference in retention time and elution sequence for daistereomers.
Bhushan and Nagar (2014b) also confirmed the elution sequence of daistereomers of β-blockers prepared with DFDNB, experimentally and also theoretically by optimising the structures of the diastereomers of Orc using the Gaussian 09 Rev A. 02 program and hybrid density functional B3LYP with 6-31G (d, p) basis set, which supported the experimental results. The mechanism in the present work is in accordance with literature (Bhushan and Nagar 2014b) that is the [(R)-L]-diastereomer retained for lesser time in comparison to that of the [(S)-L]-diastereomer which means previous one eluted first from the column.
A graphical representation for these diastereomers is shown in Fig. 4a and 4b. Fig. 4a, in [(S)-L]-diastereomer, the hydrophobic groups –CH2C6H5 on the stereogenic centre (C-1) of CDR (FDNP-L-Phe) and –C6H3(OH)2 on stereogenic centre (C-2) of Orc (in rectangle) are oriented on the same side with respect to the plane of dinitrobenzene moiety (rectangle) and are considered to be cis to each other. Fig.4b, in [(R)-L]-diastereomer, the –CH2C6H5 and –C6H3(OH)2 groups are oriented in space opposite to the plane of dinitrobenzene moiety and thus have trans-type arrangement.
In other words, the [(S)-L]-diastereomer having cis-type arrangement (Fig 4a) due to greater hydrophobicity interacts more strongly with ODS material of C18 column than the trans-type arrangement (Fig 4b) having lesser hydrophobicity, hence [(S)-L]-diastereomer has a longer retention time. Besides the different hydrophobic nature of these daistereomers, rheological properties of the mobile phase are also responsible for different partition coefficients and different retention times of [(R)-L]-and [(S)-L]-diastereomers. Therefore, the diastereomers elute one after another due to these different physical properties.
Advantages/novelty of present work
The advantages in terms of less reaction time for synthesis of diastereomers and in terms of higher Rs (for the separation of diastereomers) have been described under ‘Comparison with literature reports’. The developed CDR (FDNP-L-Phe) has high molar absorption properties with the capability of detecting diastereomers at low limits; limit of detection was found good (20 to 21 pg mL-1) for diastereomers of Orc.
Method Validation
Method validation studies were performed using diastereomers of (R,S)-Orc prepared with CDR (FDNP-L-Phe).
Linearity
The peak area response (on Y-axis) of the diastereomers of (R)-(first eluting diastereomer) and (S)-(second eluting diastereomer) enantiomer prepared with CDR were plotted against the corresponding concentration (20 to 100 ng mL-1; on X-axis). Least square method was used to complete linear regression equation. The relative standard deviation (RSD) values of slope, intercept and correlation coefficient were obtained (less than 1.4%) as a evidence of good linear relationship over this range. The regression equations were y = 0.912x –10.29 (R2 = 0.998) and y = 0.941x–11.77 (R2 = 0.999) for the diastereomers of (R)- and (S)-Orc, respectively (Table 2).
Accuracy, precision and limit of detection
Inter-day (5 days) and intra-day assay studies were carried out by replicate analysis (n = 3) of four standard solutions of diastereomeric mixtures (20, 40, 60, 100 ng mL-1) for determination of accuracy and precision. The recovery and mean SD (standard deviation) for each of the diastereomers were calculated on the basis of peak areas of first and second eluting diastereomer from the slope and intercept of the calibration plots. These are shown in Table 2. The values of relative standard deviation for [(R)-L]- and [(S)-L]-diastereomers, respectively, were 0.52% to 1.21% and 0.58% to 1.20% for intra-day precision, and 0.64% to 1.11% and 0.67% to 1.17% for inter-day precision. The recovery values for the [(R)-L]- and [(S)-L]-diastereomers were 99.81% to 101.15% and 99.35% to 101.12% for intra-day assay and 99.83% to 101.23% and 98.70% to 101.16% for inter-day assay, respectively. Limit of detection (LOD), corresponding to signal-to-noise ratio of 3, was found to be 20 pg mL-1 and 22 pg mL-1 for [(R)-L]- and [(S)-L]-diastereomers, respectively and limit of quantitation (LOQ) corresponding to signal-to-noise ratio of 10, was found to be 60 pg mL-1 and 66 pg mL-1 for [(R)-L]- and [(S)-L]-diastereomers Table 2.
CONCLUSION
The presence of strong electron withdrawing two -NO2 groups on DFDNB platform facilitate nucleophilic substitution of its F atom by amino group of chiral amino acid. While, the resonance of the lone pair of electrons of chiral auxiliaries with the -NO2 groups of the DFDNB platform resulted into high molar absorptivity of CDR, FDNP-L-Phe. Thus, the CDR so obtained is capable of detecting at concentration lower than the limit of 1% prescribed for pharmaceutical industry. It is proved that results are better in terms of resolution in comparison to certain other CSPs, CDRs and CIR reported in literature. Under MW irradiation, daistereomers synthesized with shorter reaction time in comparison to that required by other CDRs reported in literature and reaction conditions were easily optimized.
ACKNOWLEDGMENT
The authors are grateful to the DST Rajasthan, for selecting H.N. as principal investigator and other financial support to authors. Thanks are due to the Suresh Gyan Vihar University, Jaipur, for providing instrumentation and other facilities.
REFERENCES
Amini A., U. Paulsen-Sörman and D. Westerlund. 2000. Dependence of chiral separations on the amount of cyclodextrins as selectors, employing the partial filling technique in capillary zone electrophoresis. Chromatographia 51: 226–230.
Ariëns, E. J. 1984. Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology. Eur. J. Clin. Pharmacol. 26: 663–668.
Bhushan, R., and H. Brückner. 2004. Marfey’s reagent for chiral amino acid analysis: A review. Amino Acids 27: 231–247.
Bhushan, R., and H. Brückner. 2011. Use of Marfey’s reagent and analogs for chiral amino acid analysis: assessment and applications to natural products and biological systems. J. Chromatogr. B 879: 3148–3161.
Bhushan, R., and R. Kumar. 2009a. Reversed-phase high performance liquid chromatographic separation of diastereomers of α-amino alcohols and microwave assisted synthesis of Marfey’s reagent, its chiral variants and diastereomers. J. Chromatogr. A 1216: 2592–2596.
Bhushan, R., and R. Kumar. 2009b. Analysis of multicomponent mixture and simultaneous enantioresolution of proteinogenic and non-proteinogenic amino acids by reversed-phase high-performance liquid chromatography using chiral variants of Sanger’s reagent. Anal. Bioanal. Chem. 394: 1697–1705.
Bhushan, R., H. Nagar, and J. Martens. 2015. Resolution of enantiomers with both achiral and chiral phases in chromatography: conceptual challenge. RSC Adv. 5: 28316-28323.
Bhushan, R., and H. Nagar. 2013. Indirect enantioseparation of proteinogenic amino acids using naproxen-based chiral derivatizing reagent and HPLC. Biomed. Chromatogr., 27, 750-756.
Bhushan, R., and H. Nagar. 2014a. Indirect enantioseparation of selenomethionine by reversed-phase high-performance liquid chromatography using a newly synthesized chiral derivatizing reagent based on (S)-naproxen moiety. Biomed. Chromatogr., 28, 106-111.
Bhushan, R., and H. Nagar. 2014b. Enantioseparation of Orciprenaline, Betaxolol, and Propranolol using HPLC and New Chiral Reagents Based on 1,5-Difluoro-2,4-dinitrobenzene. Anal. Lett. 47: 202–219.
Bhushan, R., and H. Nagar. 2015. Resolution and isolation of enantiomers of (±)-isoxsuprine using thin layer silica gel layers impregnated with L-glutamic acid and comparison of separation of its diastereomers prepared with chiral derivatizing reagents having L-amino acids as chiral auxiliaries. Biomed. Chromatogr., 29, 357-365.
Bhushan, R., and S. Dixit. 2012. Enantioresolution of five β-blockers by reversed-phase high-performance liquid chromatography using fifteen chiral derivatizing reagents having amino acids or their amides as chiral auxiliaries on a cyanuric chloride platform. Biomed. Chromatogr. 26: 239–246.
Bhushan, R., and S. Tanwar. 2009. Reversed-phase high-performance liquid chromatographic enantioresolution of six β-blockers using dinitrophenyl-L-Pro-N-hydroxysuccinimide ester, N-succinimidyl-(S)-2-(6-methoxynaphth-2-yl) propionate and twelve variants of Sanger’s reagent as chiral derivatizing reagents. Biomed. Chromatogr. 23: 1291–1299.
Bhushan, R., V. Kumar, and S. Tanwar. 2009. Chromatographic separation of enantiomers of non-protein α-amino acids after derivatization with Marfey’s reagent and its four variants. Amino Acids 36: 571–579.
B’ Hymer C., M. Montes-Bayon, and J. A. Caruso. 2003. Marfey’s reagent: Past, present, and future uses of 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide. J. Sep. Sci. 26: 7–19.
Fujii K., Y. Ikai, T. Mayumi, H. Oka, M. Suzuki, and K. Harada. 1997. A non empirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: combination of marfey’s method with mass spectrometry and its practical application. Anal. Chem. 69: 3346–335.
ICH. ICH‐Topic Q2A: Validation of Analytical Procedures. International Conference on Harmonization of Technical Requirement for Registration of Pharmaceuticals for Human Use: Geneva, 1996.
Lee, E. J., and K. M. William. 1990. Chirality: clinical pharmacokinetic and pharmacodynamic considerations. Clin. Pharmacokinet. 18: 339–345.
Marfey, P. 1984. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg. Res. Commun. 49: 591–596.
Nagar, H. and R. Bhushan. 2014. Enantioresolution of DL-selenomethionine by: thin silica gel plates impregnated with (–)-quinine and RPTLC and HPLC separation of diastereomers prepared with difluorodinitrobenzene based reagents having L-amino acids as chiral auxiliaries. Anal. Methods, 6, 4188–4198.
Ngim, K. K., Q. Zhong, K. Mistry, and N. Chetwyn. 2012. Effect of sulfobutyl ether β-cyclodextrin modifier on selectivity of reversed phase HPLC separations. J. Liq. Chromatogr. Rel. Technol. 35: 2845–2859.
Yang, G. S., D. M. Chen, Y. Yang, B. Tang, J. J. Gao, H. Y. Aboul-Enein, and B. Koppenhoefer. 2005. Enantioseparation of some clinically used drugs by capillary electrophoresis using sulfated β-cyclodextrin as a chiral selector. Chromatographia 62: 441–445.
Zahn, H., and J. Meienhofer. 1958. Reaktionen von 1,5-difluor-2,4-dinitrobenzoyl mit insulin. I. Synthese von modellverbindungen. Makromol. Chem. Phys. 26: 126–152.
Table 1 Literature reports with present study on chromatographic separation (in terms of RS) of enantiomers of Orc using different CSPsa/CDRsb/CIRc
| S. no. | CSPsa/CDRsb/CIRc | Rs | Reference |
| 1 | Methyl-β-cyclodextrina | 3.95 | Amini et al. (2000)
|
| 2 | Sulfobutyl ether β-cyclodextrin | 0.80 | Ngim et al. (2012) |
| 3 | Cellulose tris(3,5- dimethylphenylcarbamate) a | 4.68 | Ullrich et al. (2001) |
| 4 | Sulfated-β-cyclodextrina | 2.03 | Yang et al. (2005) |
| 5 | (R)-2-(5-fluoro-2,4-dinitrophenylamino)-3-(methylthio)propanoic acidb | 9.79 | Bhushan and Nagar (2014b) |
| 6 | (S)-N-(1-cyclohexylethyl)-5-fluoro-2,4-dinitrobenzenamineb | 10.22 | Bhushan and Nagar (2014b) |
| 7 | (R)-5-fluoro-2,4-dinitro-N-(1-phenylethyl)benzenamineb | 8.26 | Bhushan and Nagar (2014b) |
| 8 | L-Glutamic acidc | 2.10 | Bhushan et al. (2015) |
Table 2 Summary of HPLC method validation containing linearity, intra‐ and inter‐day precision, and recovery studies
| First eluting diastereomer | Second eluting diastereomer | ||||||||||
| Linearity | |||||||||||
| Range | 20 to100 ng mL-1 | 20 to 100 ng mL-1 | |||||||||
| Slope | 0.912 | 0.941 | |||||||||
| Intercept | –10.29 | –11.77 | |||||||||
| Correlation Coefficient (R2) | 0.998 | 0.999 | |||||||||
| Actual concentration
ng mL-1 |
Mean±SD (measured)
ng mL-1 |
Recovery (%)
|
RSD (%)
|
Mean±SD (measured)
ng mL-1 |
Recovery (%)
|
RSD (%)
|
|||||
| Intra-day precision (n = 3) | |||||||||||
| 20 | 10.109 ± 0.122 | 99.93 | 1.21 | 10.116 ± 0.131 | 99.97 | 1.20 | |||||
| 40 | 19.843 ± 0.175 | 101.105 | 0.88 | 19.789 ± 0.162 | 101.12 | 0.82 | |||||
| 60 | 30.116 ± 0.174 | 99.81 | 0.58 | 30.215 ± 0.195 | 99.35 | 0.65 | |||||
| 100 | 49.450 ± 0.255 | 101.05 | 0.52 | 49.612± 0.288 | 101.08 | 0.58 | |||||
| Inter-day precision (n = 3) | |||||||||||
| 20 | 9.929 ± 0.110 | 100.05 | 1.11 | 10.168 ± 0.118 | 101.16 | 1.17 | |||||
| 40 | 19.956 ± 0.196 | 99.89 | 0.98 | 19.923 ± 0.189 | 99.96 | 0.95 | |||||
| 60 | 30.158 ± 0.236 | 99.83 | 0.75 | 30.105 ± 0.217 | 100.09 | 0.72 | |||||
| 100 | 50.853 ± 0.325 | 101.23 | 0.64 | 49.790 ± 0.334 | 98.70 | 0.67 | |||||
| Sensitivity | |||||||||||
| Limit of detection (pg mL-1) | 20 | 22 | |||||||||
| Limit of quantification (pg mL-1) | 60 | 66 | |||||||||
Figure Captions
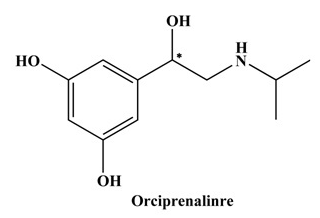
Fig. 1 Structures of Orciprenaline (* represents chiral centre).
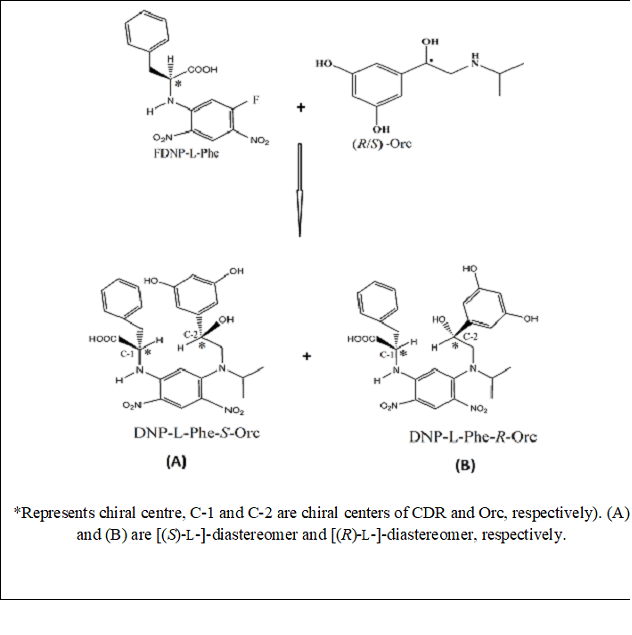
Fig.2 Scheme for synthesis of daistereomers of (R/S)-Orc with CDR (FDNP-L-Phe)
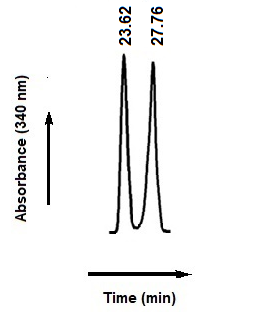
Fig. 3 Sections of chromatograms showing resolution of diastereomers of Orc prepared with CDR (FDNP-L-Phe).
Chromatographic conditions: Promosil C18 (250 × 4.6mm i.d., 5 µm particle size); mobile phase 2, linear gradient (30 to 65%) of MeCN with 0.10% TFA for diastereomers of Orc in 45min; flow rate, 1.0mL/min; detection, 340 nm. From left to right, first and second peaks are for [(R)-L]- and [(S)-L]-diastereomers, respectively.
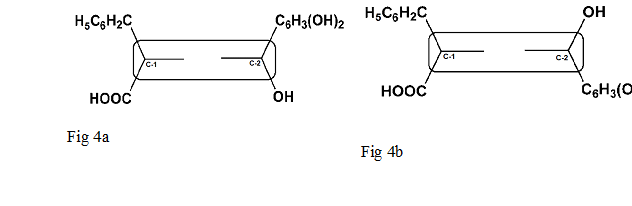
Fig. 4 In [(S)-L]-diastereomer, the hydrophobic groups, -CH2C6H5 on the stereogenic centre of CDR (C-1) and -C6H3(OH)2 on stereogenic centre of Orc (C-2), are appearing on the same side with respect to the plane of dinitrobenzene moiety and thus the two groups are oriented in a cis-type arrangement (Fig. 4a). In [(R)-L]-diastereomer, the -CH2C6H5 and -C6H3(OH)2 groups are oriented, in space, opposite to the plane of dinitrobenzene moiety and thus have trans-type arrangement (Fig.4b).